
Роботу виконали:
Міщенко Андрій Володимирович,
учень 10-А класу
Кременчуцького ліцею № 4 «Кремінь»
Кременчуцької міської ради
Полтавської області
Кіндра Максим Олексійович,
студент І курсу хімічного факультету Інституту високих технологій
Київського національного університету імені Тараса Шевченка
Науковий керівник:
Джавахішвілі Сергій Георгійович,
заступник директора з навчально-виховної роботи
Кременчуцького ліцею № 4 «Кремінь»
Кременчуцької міської ради
Полтавської області.
переможець V Всеукраїнського Інтернет-конкурсу
„УЧИТЕЛЬ РОКУ – 2020” за версією науково-популярного
природничого журналу „КОЛОСОК” у номінації „Хімія”
Вступ
Основою великої кількості сучасних технології є матеріали, до яких відносяться напівпровідники, феромагнетики, п’єзоелектрики, чутливі до ультрафіолетового та інфрачервоного випромінювання кристали, кристалічні плівки для мікроелектроніки та комп’ютерної індустрії. Виробництво кожного з цих матеріалів включає етапи вирощування кристалів. Сьогодні кристали можна однозначно визначити як «фундаментальні стовпи» сучасних передових технологій, які відіграли важливу роль у багатьох технологічних новинках ХХ століття. Масштаби виробництва кристалів та пристроїв на їх основі постійно зростають та обумовлені широким попитом людства відповідних товарів. Величезна кількість предметів побуту, використання яких сьогодні є доволі звичним для кожної людини, не були б можливі без кристалів. Радіоприймачі, телевізори, програвачі, слухові апарати, автомобільні системи запалювання і багато-багато інших пристроїв містять кристалічні компоненти [1].
Можливість приготування великих монокристалів неорганічних та органічних сполук є важливим фактором у багатьох галузях науки. Маючи «великий» монокристал, науковець отримує можливість достатньо точно описати міжатомні відстані у хімічному зв’язку та особливості координаційної сфери атомів за допомогою методів рентгеноструктурного аналізу (РСА) або в умовах нейтронного опромінення. Використання кристалів часто ґрунтується і на їх фізичних властивостях: п’єзоелектрична природа кварцу, кристали якого здатні генерувати електричний струм при деформації, або напівпровідникові властивості арсенідів Силіцію, Германію та Галію [2].
Окремою галуззю науки за останні десятиріччя стала хімія рідких кристалів. Рідкокристалічний стан речовини виникає через існування нековалентних, залежних від взаємної орієнтації у просторі взаємодій між молекулами у конденсованій фазі. Через оборотність процесів, що визначаються рівновагою внутрішньо- та міжмолекулярних сил, які сприяють утворенню рідких кристалів, цей стан речовини може бути викликаний, в основному, тільки за умов стимуляції ззовні (наприклад, електромагнітне поле у дисплеях телевізорів, мобільних телефонів тощо) [3]. Явище існування четвертого агрегатного стану речовини «мезофази» (цей термін, введений німецьким фізиком Отто Леманом, буквально означає «проміжний стан»), відкрите у 1888 році австрійським ученим-ботаніком Фрідріхом Рейнітцерем, вже за сто років зробило справжню революцію у розвитку технологій.
Існує декілька методів вирощування великих монокристалів неорганічних та органічних сполук. Вирощування кристалу із водного розчину солі є часто першим досвідом наукової діяльності юного хіміка. Серед найбільш поширених методологій утворення великих монокристалів можна виділити [2]:
- кристалізація із насиченого водного розчину при охолодженні або випаровуванні;
- потоковий (flux) метод;
- електролітичний метод;
- гідротермальний синтез;
- гельовий метод;
- метод Czochralski, що оснований на буквальному витягуванні стержнів кристалів із розплавів солей (наприклад, кальцій вольфрамат);
- методи, що включать використання газової фази (сублімація) тощо.
Одним із найпростіших, але ще недостатньо вивчених, методів вирощування кристалів є технологія використання гелю [4]. Для розуміння процесів, що відбуваються під час такого експерименту, простого з точки зору методики проведення, необхідно мати ґрунтовні знання в області властивостей гелів та кристалів, структурних особливостей цих матеріалів. Тим не менш, такий експеримент дозволить пояснити достатньо складні феномени синерезису або нуклеації. Одночасно з цим, такі, специфічні для гелів, явища як «Оствальдівська перекристалізація» [5] та «кільця Лізеганга» [6] (Рис. 1) набули широкого використання у виробництві штучного напівдорогоцінного каміння та біжутерії.
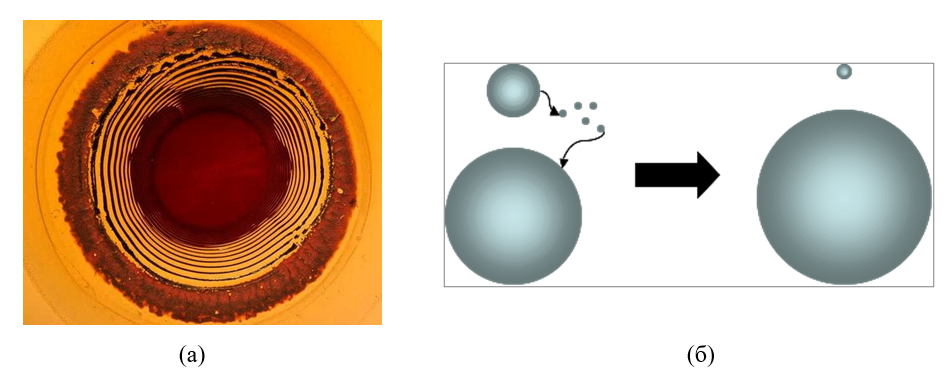
Рис. 1. Приклад «кілець Лізеганга», утворених осадом арґентум(I)дихромату у шарі желатинового гелю (а) та схема процесу Оствальдівської перекристалізації
Основною метою нашої роботи стало дослідження кінетики росту кристалів міді під час відновлення йонів Cu2+ із водного розчину купрум(II)сульфату металічним алюмінієм в гелі агару та, досліджуючи структурні особливості отриманих кристалів, довести гіпотезу можливого механізму проходження процесу шляхом утворення мідно-алюмінієвого гальванічного мікроелементу.
Розділ 1. Основна частина
1.1. Гелі як середовище для хімічних процесів
Гель можна визначити як двокомпонентна система, один із компонентів якої утворює тривимірну макромолекулярну структуру, що виступає в якості каркасу, пустоти якого заповнені низькомолекулярним розчинником, дисперсійним середовищем. Серед індивідуальних речовин та сумішей, здатних утворювати гідрогелі (дисперсійним середовищем виступає вода), можна виділити полісилікатну кислоту, олеати, желатин, поліетиленгліколь, агар-агар.
Хоча гелі мають вигляд та проявляють властивості твердого агрегатного стану, швидкість дифузії низькомолекулярних речовин в розведених студнях майже не відрізняється від вільної дифузії в розчинах, поза як масова частка води в них може досягати 99%, а відсутність текучості обумовлена не високою в’язкістю, а сітчастою структурою високомолекулярного каркасу. Рухливість невеликих за розмірів іонів у гелі також мало відрізняється від рухливості у розчині. Тому електропровідність розчинів майже не змінюється при гелеутворенні [7].
Основною відмінністю гелів від розчинів є відсутність конвекції. Тому деякі процеси в них мають свої особливості, зокрема специфічно проходять реакції із утворенням малорозчинних речовин. Так, наприклад, якщо в гель желатину завчасно внести деяку кількість калій дихромату, а на студень нашарувати більш концентрований розчин арґентум(I)нітрату, то на межі контакту розчинів одразу ж утвориться тонкий шар осаду арґентум(I)дихромату. Через деякий час в результаті дифузії катіонів Ag+ осад поширюється всередину гелю, але не суцільною масою: виникають періодичні зони осадження, що перемежовуються із прошарками чистого гелю. Ритмічні, шарові відкладення осадів в гелях вперше були описані німецьким хіміком Рафаелем Лізегангом та названі на його честь: «кільця Лізеганга». Періодичними реакціями пояснюється шаровий розподіл забарвлення багатьох мінералів: яшми, малахіту, агату тощо.

Рис. 1.1. Приклади утворення кілець Лізеганга (а) в процесі осадження магній гідроксиду Mg(OH)2 із желатинового гелю магній сульфату MgSO4 амоній гідроксидом NH4OH та (б) арґентум(I)дихромату Ag2Cr2O7 із силікатного гелю калій дихромату K2Cr2O7 аргентум(I)нітратом AgNO3.
Важливими факторами, що визначають перебіг процесів, які супроводжують проведення хімічних реакцій або вирощування кристалів в гелях є:
- кислотність середовища (для різних типів гелів різні значення рН можуть як запобігати, так і сприяти його миттєвому утворенню. Саме тому кислотність середовища має постійно відслідковуватися за допомогою рН-метру протягом усього часу проведення експерименту в гелі);
- концентрація реагентів (визначаються для кожної системи окремо);
- вибір середовища та послідовність розчинення реагентів (визначається експериментально. Зазвичай один із реагентів розчиняють ще до формування гелю);
- температура нуклеації або кристалізації;
- форма та розмір посудини, що використовуватиметься при проведенні експерименту.
Останній фактор є часто визначальним з точки зору швидкості росту кристалів: чим повільніше відбуватиметься процес, тим більшим буде утворений монокристал. Так, для формування великих монокристалів солей використовують U-трубки [2]. Типова методика використання U-трубки для такого експерименту полягає в попередньому утворенні гелю в такому реакторі та додавання розчинів двох реагентів в протилежних кінцях U-трубки. Зустрічна дифузія йонів або молекул реагуючих речовин один до одного приводить до росту великих монокристалів. Змінюючи або підтримуючи концентрацію реагентів сталою, можна досягти вражаюче великих розмірів монокристалів [4].
Таким чином, використання гелю в якості реакційного середовища дозволяє не тільки краще візуалізувати перебіг хімічного процесу, вповільнюючи доставку реагентів один до одного, а й, як результат, покращити фізичні характеристики отримуваних таким чином кристалів, що дозволить проектувати нові джерела енергії, органічні напівпровідники, надтонкі рідкокристалічні плівки, лазери, високотемпературні надпровідники тощо.
1.2. Алюмінієва корозія та швидкість реакції
Завдяки таким властивостям, як мала густина, висока тепло- і електропровідність, висока пластичність, достатньо високі міцнісні властивості (особливо в сплавах) і багатьом іншим цінним властивостям алюміній отримав винятково широке розповсюдження в різноманітних галузях сучасної техніки і відіграє найважливішу роль серед кольорових металів. Важливою особливістю застосування алюмінію в техніці є те, що він досить складно піддається пайці та лудінню. Хімічно стійка оксидна плівка, утворювана на його поверхні, важко видаляється за допомогою звичайних флюсів.
З огляду на це, важливою промисловою проблемою є алюмінієва корозія. Численні дослідження показали, що окисне руйнування алюмінію значно пришвидшується у морській воді або в контакті з міддю, як металевою, так і в розчині Cu2+:
- Фактор морської води пояснюється так званим ефектом «хлоридного прискорення». Однак в літературі відсутні ґрунтовні дослідження щодо порівняння ефекту аніонів Cl‑ із впливом інших галогенід-йонів.
- Другий фактор обумовлений великою різницею електродних потенціалів мідного (ECu2+/Cu>0) та алюмінієвого (EAl3+/Al<0) електродів. Позитивне значення електрорушійної сили утвореного таким чином гальванічного мідно-алюмінієвого елемента сприяє самочинному перебігу процесу окиснення металічного алюмінію.
- Окремим важливим моментом алюмінієвої корозії у водному розчині є вплив кислотності:
Al2O3 + 6H3O+ 3H2O → 2[Al(H2O)6]3+, (pH<4)
Al2O3 + (2x-6)OH‑ + (15-2x)H2O→ 2[Al(OH)x(H2O)6-x]3-x, (pH>9)
Утворений під впливом агресивного навколишнього середовища міцний, непроникний шар алюміній оксиду Al2O3 може руйнуватися в умовах як кислого розчину (рН<4), так і лужного (рН>9), прискорюючи алюмінієву корозію.
У нашій роботі ми спробували оцінити вплив кожного із цих факторів на швидкість перебігу реакції заміщення міді алюмінієм.
Реакція заміщення міді алюмінієм із розчину відповідної солі Купруму часто демонструється в навчальному процесі в якості класичного прикладу «простої» окисно-відновної реакції, а також є чудовою ілюстрацією електрохімічного ряду напруги металів. Виконання такого експерименту в ході лабораторного практикуму дозволяє встановити та пояснити емпіричну формулу CuCl2 [8].
У той же час просте, на перший погляд, заміщення атомів одного металу іншим є достатньо складним процесом [9-10] вже хоча б через те, що хімічна реакція відбувається на межі двох фаз, і не може бути описана стандартним кінетичним рівнянням першого порядку гомогенних реакцій:
2Alтв + 3[Cu(H2O)4]2+ → 2[Al(H2O)6]3+ + 3Cuтв
Загальноприйнятим є механізм перебігу такої реакції, що включає п’ять стадій:
- дифузія йонів Cu2+ до поверхні металічного алюмінію;
- адсорбція йонів Cu2+;
- електронний перехід від Al до Cu2+;
- десорбція утворених йонів Al3+;
- дифузія йонів Al3+ від поверхні металу в розчин.
Зазвичай, стадії (1) та (3) є швидкість визначальними. Процеси, що мають передувати зазначеним вище (1)-(5) та які необхідно врахувати (розчинення оксидної плівки Al2O3; нуклеація міді та збільшення шорсткості поверхні металу), повністю залежать від доступної площі алюмінієвої поверхні. Ці, передуючі основним стадіям реакції, процеси обумовлюють так званий час індукції, протягом якого швидкість росту кристалів міді є меншою. Інтегральна форма кінетичного рівняння такої реакції матиме вид [12]:
![]()
де А – величина площі поверхні алюмінію, θ0 – частина поверхні алюмінію невкрита шаром оксиду Al2O3, V – об’єм розчину солі Cu2+, tind – час індукції. Коли t велике, вираз [1 – exp(-t/tind)] стає рівним одиниці. За умови, що алюмінієва стрічка жодним чином попередньо не оброблялася, під впливом агресивного середовища повітря її поверхня майже повністю вкрита поверхневим шаром оксиду Al2O3, що дає повне право зробити припущення про значення θ0=0. Враховуючи ці припущення інтегральна форма кінетичного рівняння відповідатиме рівнянню реакції першого порядку та матиме вигляд:
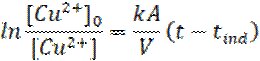
Експериментальне визначення залежності ln([Cu2+]0/[Cu2+]) порівняно з t(хв.) протягом усього часу перебігу реакції має дати вид прямої, кут нахилу якої відповідатиме kA/V, а перетин із віссю абсцис – час індукції tind (хв.).
З метою виявлення ефекту впливу сторонніх йонів на швидкість хімічної реакції ми провели цикл експериментів із використанням розчинів солей купрум(II)сульфату CuSO4 (100 мл 0.2М CuSO4٠5H2O), купрум(II)хлориду CuCl2 (50 мл 0.2М CuCl2٠2H2O) та купрум(II)броміду CuBr2 (50 мл 0.2М CuBr2) при температурі 20°С.
Протягом усього часу реакції кожну хвилину за допомогою піпетки відбирали пробу та аналізували за допомогою спектрофотометру КФК-3 (фильтр UV-vis, 500-900 нм). Припустивши, що оптична густина розчину обумовлена виключно забарвленням комплексних йонів [Cu(H2O)4]2+, відповідно до закону світлопоглинання Бугера-Ламберта-Бера, інтегральну форму кінетичного рівняння можна записати так:
![]()
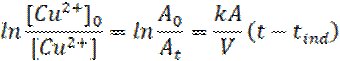
де А0 та Аt – оптична густина розчинів на початку реакції та в моменту часу t(хв.).
Як результат, для реакцій за участю всіх трьох солей Cu2+ отримані лінійні залежності (Рис. 1.2.1 та 1.2.2).
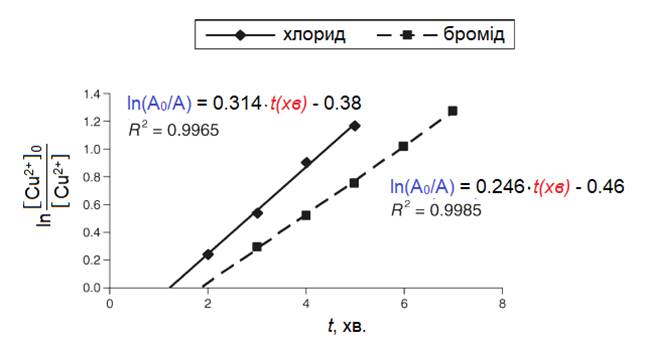
Рис. 1.2.1. Кінетичні дослідження реакції алюмінію із розчинами CuCl2 та СuBr2
Аналіз отриманих даних щодо реакцій галогенідів із алюмінієвою стрічкою показав, що вони дають однаковий вид кінетичних прямих, але реакція за участю купрум(II)броміду є трохи повільнішою ({kA/V}Cl=0,314 хв-1, а {kA/V}Br=0,246 хв-1) і має більший час індукції (tind(CuCl2)=1,22 хв., а tind(CuBr2)=1,87 хв.). Виконати аналогічне дослідження для купрум(II)йодиду неможливо, оскільки CuI2 є нестійким і самочинно розкладається, утворюючи купрум(I)йодид та молекулярний йод.
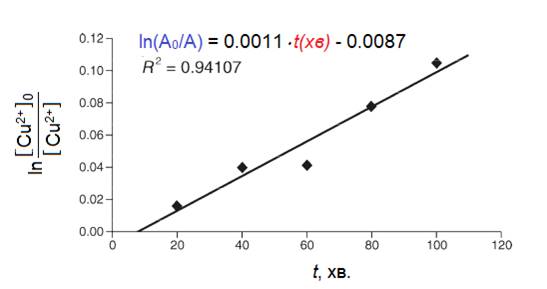
Рис. 1.2.2. Кінетичні дослідження реакції алюмінію із розчином CuSO4
Порівняно із галогенідами реакція за участю купрум(II)сульфату (рис. 1.2.2) відбувається приблизно у 100 разів повільніше (час індукції tind(CuSO4)=7,91 хв.): приводячи до однакових об’ємів розчинів (Vрозч.(CuSO4)=2٠Vрозч.(CuX2)), отримуємо відношення приведених констант швидкості реакцій
k(CuCl2):k(CuSO4)=143:1,
k(CuBr2):k(CuSO4)=112:1,
що однозначно дозволяє зробити висновок про існування прискорюючого ефекту корозії алюмінію не просто хлоридами Cl‑, а в цілому галогенідами (Х‑). Такий вплив можна пояснити можливістю невеликих за розміром галогенід-аніонів утворювати тетраедричні комплекси [AlX4]‑ з катіонами Al3+. До того ж є чітка кореляція між константою комплексоутворення (lgK(AlCl4‑)>lgK(AlBr4‑)) та впливом аніону на швидкість перебігу реакції: Cl‑>Br‑. Аналіз літературних даних [11] показав, що при досягненні критичної концентрації Cl‑ у розчині на поверхні алюмінію, галогенід аніони можуть проникати в структуру алюміній оксиду через невелику різницю між йонними радіусами О2‑ та Cl‑ (1.3Ȧ та 1.4Ȧ відповідно), стимулюючи процес розчинення оксиду:
Al(O)OH + H3O+ + mCl‑ + (5-n)H2O ‑ 3e‑ → [AlCln(OH)2(Н2O)4-n]1-n + (m-n)Cl‑
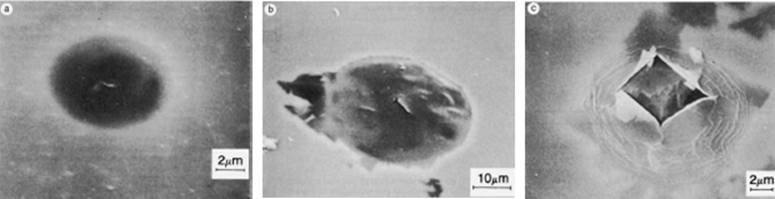
Рис. 1.2.3 Утворення блістер у поверхневому оксидному шарі металічного алюмінію та їх руйнування в процесі виділення газуватого водню [11].
Електрони, що віддаються атомами металу та накопичуються у поверхневому оксидному шарі, приєднується катіонами Н3О+ з утворенням газуватого водню (Рис. 1.2.3). Виділення Н2 під плівкою спричиняє утворення характерних блістер, які із досягненням критичного значення внутрішнього тиску розриваються, обумовлюючи руйнування оксидного шару та втрату металом можливості опиратися корозійним процесам [11].
Додатковим фактором активації поверхні металу є природна кислотність розчинів Cu2+ (pH»4):
Cu2+ + 2Н2О → Cu(ОН)+ + Н3О+
Як вже загадувалось вище оксидний поверхневий шар Al2O3 спонтанно розчиняється в розчинах із рН<4. Для перевірки можливого впливу рН розчинів галогенідів CuX2 та сульфату CuSO4 на швидкість реакції ми виміряли рН кожного вихідного сольового розчину Купруму(II): значення рН(0,2М CuCl2)=3.4, а рН(0,2М CuSO4)=3.9. Не зважаючи на незначні відмінності обидва розчини мають рН<4, що відповідає порогу самочинного перебігу розчинення Al2O3.
Таким чином, можна зробити висновок щодо впливу на корозію алюмінію кислотності середовища, як не домінуючого фактору в присутності галогенід-йонів. Крапку в цьому питанні можуть поставити серія експериментів в розчинах із суттєво різним значенням рН, що досягається шляхом використання буферних розчинів.
1.3. Кінетика росту кристалів міді
Наступним етапом нашої роботи стало вивчення характеру росту кристалів міді в процесі відновлення їх металічним алюмінієм у 2%-му агар-агаровому гелі. З метою депасивації захисного оксидного шару металічного алюмінію в якості реакційного середовища використовували гель, що містив 0,1 М хлорид-аніонів Cl‑.
Дослідження кінетики росту кристалів міді на поверхні алюмінію в процесі реакції відновлення катіонів Cu2+ потребує створення умов для їх рівномірного росту. Саме тому експеримент проводили в гелі агару. Основними причинами вибору такого реакційного середовища є:
- порівнюючи з іншими гелеутворювачами, приготування гелю агару є зручним та не потребує багато часу;
- гель агару запобігає шкідливому для росту кристалів міді впливу бульбашок водню (Рис. 1.3.1);
- гель агару є досить прозорим, що полегшує спостереження за процесами.
Підвищена кислотність розчину Cu2+ та висока активність алюмінію (Е0 = ‑1.70 В) спричиняє активне виділення бульбашок водню (Рис. 1.3.1). Для нівелювання їх впливу на спостереження за ростом кристалів міді обов’язковим є досягнення моменту повноцінного утворення гелю.
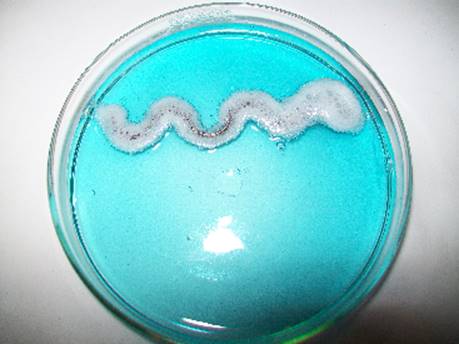
Рис. 1.3.1. Виділення водню на поверхні алюмінієвої пластинки у водному розчині солі купрум(II)сульфату.
Зростання кристалів міді (Рис. 1.3.2) з поверхні алюмінієвої пластинки в оточуючий її гель має майже лінійний характер. Видно, що впродовж експерименту колір гелю на відстані близькій до поверхні пластинки (на початку) та гілок міді (впродовж росту кристалів) є світлішим, що вказує на постійне «поглинання» гідратованих катіонів [Cu(H2O)4]2+ внаслідок реакції.
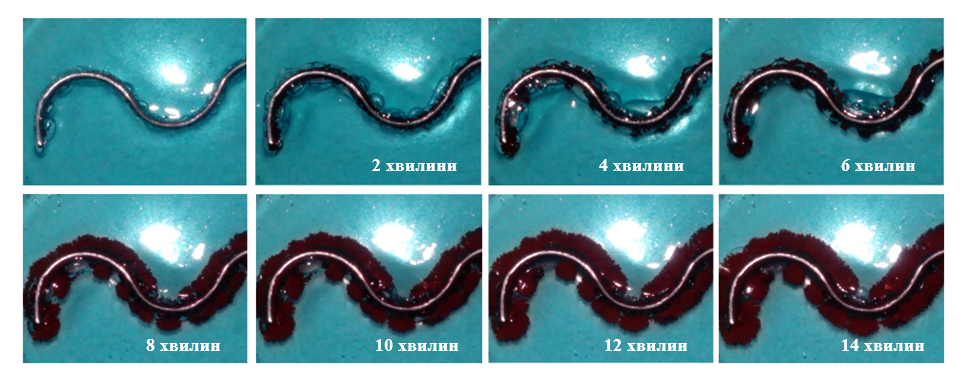
Рис. 1.3.2. Зростання кристалів міді в оточуючий алюміній гель агару.
Як результат, нами отримана залежність (Рис. 1.3.3) збільшення довжини (мм) окремої «гілки» мідного кристалу з часом (хв.):
l (мм) = 0.303t + 1.426(1‑e‑0.387t)
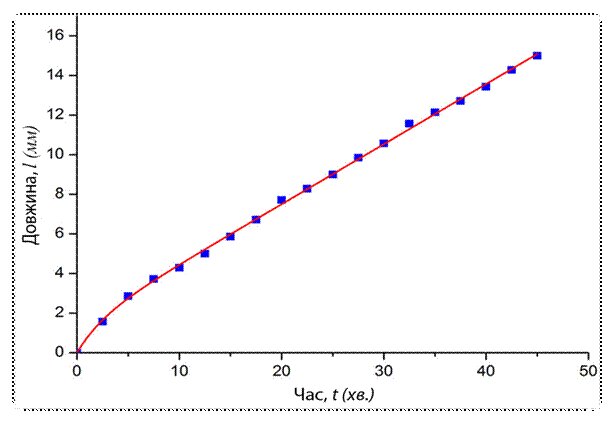
Рис. 1.3.3. Графік збільшення росту l (мм) кристалу «гілки» міді з часом t (хв.)
На перший погляд, близький до лінійного характер росту кристалу суперечить загальним уявленням про перебіг хімічного перетворення. Загальний вигляд кінетичного рівняння реакції по відношенню до зміни концентрації кожного з учасників процесу включає в себе рівноважні концентрації гідратованих катіонів Cu2+ та металічного алюмінію:
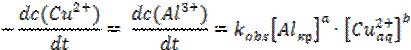
Тобто, зменшення концентрація реагентів має вповільнювати реакцію. Концентрація реагуючої частинки можна визначити як імовірність її участі в елементарному акті швидкість визначальної стадії процесу. Оскільки реакція відбувається на поверхні металу, його концентрацію, що визначається питомою активною поверхнею, можна прийняти рівною одиниці [Alкр]=1 моль/л, а висока рухливість іонів в 2%-му гелі агару та постійна дифузія Cu2+ до поверхні металу забезпечує їх сталу концентрацію протягом тривалого часу (50 хвилин) [Cuaq2+]=const. Диференціальна форма кінетичного рівняння в такому випадку матиме вид:
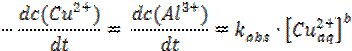
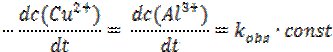
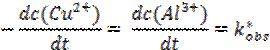
де k*obs – приведена константа швидкості реакції.
1.4. Мідно-алюмінієвий гальванічний мікроелемент
Досліджуючи будову кристалів «гілок» міді на різних етапах їх росту, можна побачити, що осадження міді відбувається не на поверхні алюмінієвої пластинки, а на вже утворених мідних кристалах (Рис. 1.4.1). Цей висновок чудово узгоджується із отриманою «лінійною» залежністю росту кристалів (Рис. 1.3.3): оскільки катіони Cu2+ відновлюються безпосередньо на поверхні зростаючих в гель мідних кристалів, їх концентрація залишається сталою та рівною початковій.
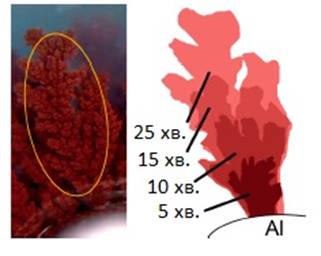
Рис. 1.4.1. Зміна форми кристалу міді під час перебігу хімічної реакції (збільшення в ≈ 100 разів)
З метою пояснення факту росту кристалів на поверхні міді, ми висунули гіпотезу утворення гальванічних мікроелементів між алюмінієвим анодом та мідним катодом. Анодний процес відбувається на поверхні алюмінієвої пластинки та приводить до поступового розчинення металу (Al – 3e‑ → Al3+, Е0Al/Al3+ = –1.663 В). На катоді можливе одночасне проходження двох процесів: відновлення міді (Cu2+ + 2e‑ → Cu, Е0Cu/Cu2+ = +0.337 В) та водню (2H3O+ + 2e‑ → H2 + 2H2O).
Виникнення гальванічної пари відбувається при безпосередньому контакті алюмінію на міді, до того ж роль зростаючих «гілок» міді можна визначити як провідників, якими електрони рухаються від алюмінієвої пластинки до мідного кристалу. «Двигуном» такого окисно-відновного процесу є електрорушійна сила, що виникає між двома електродами:
![]()
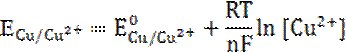
![]()
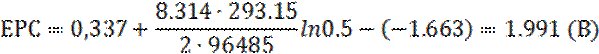
(‑) Al | 0.1M Cl– | 0.5M Cu2+ | Cu (+)
Рис. 1.4.2. Схема (контактна) моделювання утворення гальванічного елемента

Рис. 1.4.3. Результат перебігу реакції в умовах моделювання утворення гальванічного елементу.
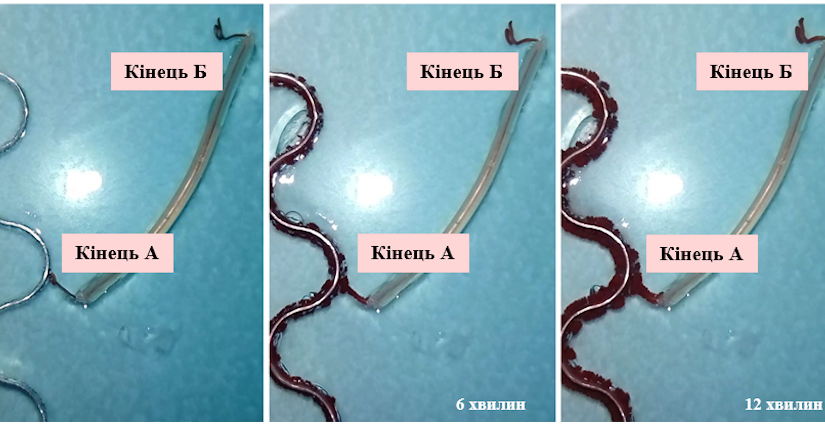
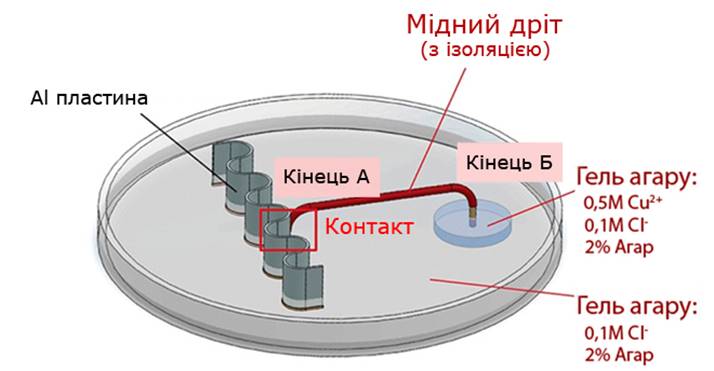
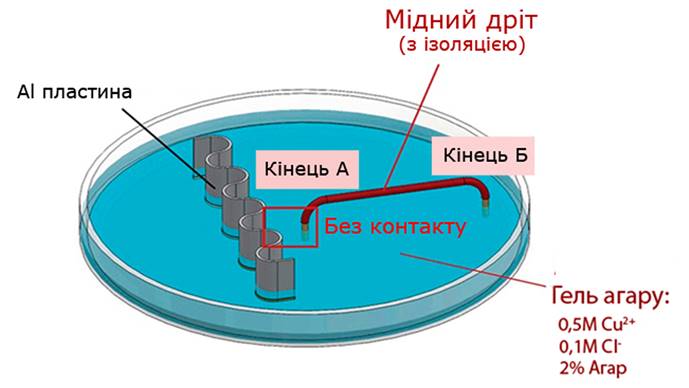
Для підтвердження висунутої гіпотези ми провели експеримент, в умовах якого алюмінієву пластинку помістили в гель агару, що містив тільки 0.1М Cl‑. Один кінець мідного дроту із ізоляційним покриттям прикріпили до пластинки (кінець А), а інший опустили в гель (кінець Б), що містив 0.5М Cu2+ (Рис. 1.4.2.). Як результат, можна спостерігати утворення мідних кристалів виключно на кінці Б (Рис. 1.4.3).
(‑) Al | 0.1M Cl‑, 0.5M Cu2+ | Cu (+)
Рис. 1.4.4. Схема (безконтактна) моделювання утворення гальванічного елемента
Правда, цей експеримент моделює утворення гальванічного елемента, отриманого із мідного та алюмінієвого електродів, які знаходяться в розчинах різних електролітів (мідний катод в розчині Cu2+ та алюмінієвий анод в розчині Cl‑), і не доводить однозначно його існування в умовах реакції заміщення.
Остаточну крапку в питанні виникнення гальванічної пари поставив експеримент, в ході якого алюмінієву пластинку та мідний дріт помістили в один і той же гель із 0.5М Cu2+та 0.1М Cl‑ на деякій маленькій відстані один від одного: кінець А мідного дроту не торкається поверхні металу (Рис. 1.4.4).
Як результат, відновлення Cu2+ розпочалось на поверхні алюмінієвої пластинки. Але при досягненні зростаючих мідних кристалів кінця А мідного дроту осадження міді можна спостерігати і на іншому його кінці Б (Рис. 1.4.5), що однозначно доводить виникнення мідно-алюмінієвих гальванічних мікроелементів.
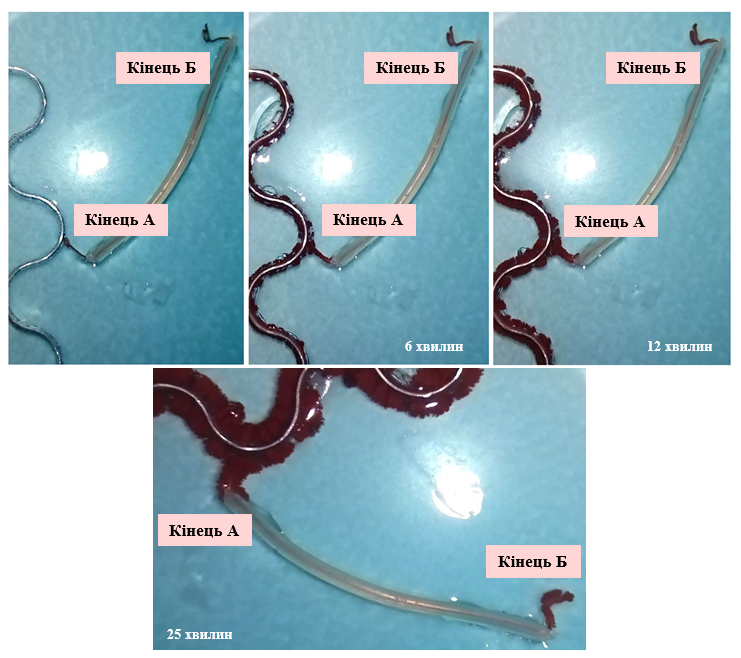
Рис. 1.4.5. Результат перебігу реакції в умовах моделювання утворення гальванічного елементу
Таким чином можна зробити остаточний висновок про роль утворюваних «гілок» міді як провідників між парою електродів: алюмінієвим анодом, окиснення якого спричиняє поступове його розчинення в гелі агару, та мідним катодом, що представляє собою постійно зростаючі в напрямку гелю мідні кристали.
Розділ 2. Експериментальна частина
Усі реагенти (CuSO4∙5H2O, CuCl2∙2H2O, CuBr2, NaCl, агар, алюмінієва пластина) були отримані із комерційних джерел та використовувались без додаткової очистки. Усі процеси, що супроводжують перебіг хімічної реакції були записані за допомогою цифрової 13Мпкс камери смартфону Lenovo P70А.
УТВОРЕННЯ ГЕЛЮ 0.5М Cu2+/0.1M Cl‑
До 100 г нагрітої до 900С дистильованої води додали 2 г сухого агару. Після повного розчинення агару при постійному перемішуванні всипали 12.5 г мідного купоросу (0.05 моль) та 0.6 г натрій хлориду (0.01 моль). Утворений таким чином розчин розлили у декілька чашок Петрі (товщина шару гелю склала 3-5 мм). При охолодженні розчинів до 450С-500С почалось формування гелю.
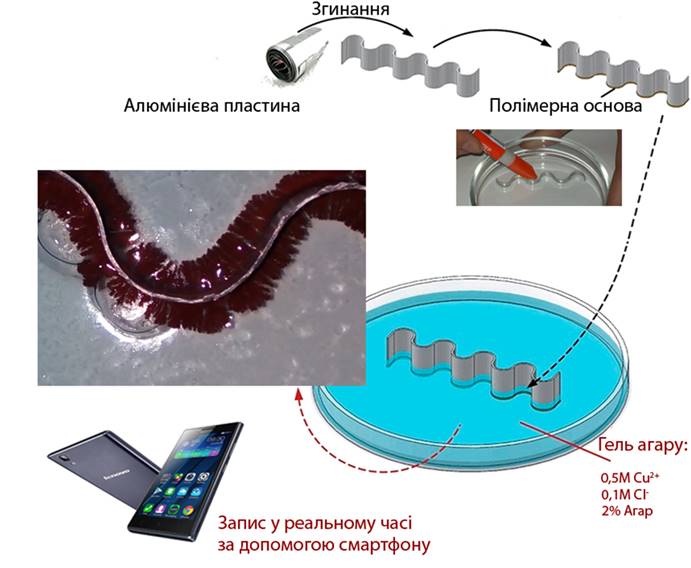
Рис. 2.1. Схема методики проведення експерименту.
УТВОРЕННЯ ГЕЛЮ 0.1M Cl‑ ІЗ «ОСТРОВОМ» 0.5М Cu2+/0.1M Cl‑
До 100 г нагрітої до 900С дистильованої води додали 2 г сухого агару. Після повного розчинення агару при постійному перемішуванні висипали 0.6 г натрій хлориду (0.01 моль). Утворений таким чином розчин розлили у декілька чашок Петрі (товщина шару гелю склала 3-5 мм). При охолодженні розчинів до 450С-500С почалось формування гелю.
У добутий гель помістили алюмінієву пластинку (товщиною 0.5 мм). На відстані 3-5 см від поверхні металу за допомогою циліндричної пробірки видалили частину гелю та заповнили утворений простір гарячим гелем із 0.5М Cu2+/0.1M Cl‑ (Рис. 2.2).

Рис. 2.2. Утворення гелю 0.1M Cl‑ із «островом» 0.5М Cu2+/0.1M Cl‑.
Висновки
Просте заміщення атомів одного металу іншим із розчину відповідної солі є достатньо складним процесом, особливо, якщо мова йде про метал, поверхня якого значно пасивована утвореною оксидною плівкою.
Роль сторонніх факторів, таких як кислотність середовища або наявність іонів, що, на перший погляд, не приймають участь в перетворенні може бути навіть визначальною. Аналіз даних, отриманих в результаті серії проведених експериментів по вивченню впливу таких «сторонніх» йонів, змушує зробити висновок щодо їх безпосередньої участі в процесі корозії поверхні алюмінію, значно прискорюючи перебіг хімічної реакції.
Дослідження процесу відновлення катіонів Cu2+ металічним алюмінієм в присутності аніонів Cl‑ в гелі агару виявило лінійний характер росту кристалів міді, що чудово узгоджується зі знаннями про дифузійні процеси в гелях. Детальне вивчення особливостей зміни структури зростаючих в гель протягом реакції «гілок» міді також є підтвердженням спостережуваної залежності: осадження атомів Купруму відбувається на поверхні вже утворених кристалів, а не на поверхні алюмінієвої пластинки. Для пояснення цього факту висунута гіпотеза утворення в процесі реакції мідно-алюмінієвих мікроелементів. Проведені експерименти за участю такої гальванічної пари в гелі агару в умовах реакції заміщення стали яскравим доказом можливості такого перебігу процесу та дали змогу однозначно визначити роль «гілок» мідного кристалу, як провідника між анодом (поверхня металічного алюмінію) та катодом (поверхня металічної міді).
Список використаних джерел
- Mroczkowski S. Needs and Opportunities in Crystal Growth / S. Mroczkowski // J: Chem. Educ.‑ 1980.‑ 57(8).‑ P. 537-541.
- Suib S. L. Crystal Growth in Gels / S. L. Suib // J: Chem. Educ.‑ 1985.‑ 62(1).‑ P. 81-82.
- Carlton R. J. Chemical and biological sensing using liquis crystals / R. J. Carlton, J. T. Hunter, D. S. Miller, R. Abbasi, P. C. Mushenheim, L. N. Tan, N. L. Abbott // J. Crystals Rev.‑ 2013.‑ 1(1).‑ P. 29-51
- Henisch H. K. Crystal Growth in Gels / H. K. Henisch // The Pennsylvania State University Press: University Park, PA.‑ 1973.
- а) Ostwald W. D. / Z. Phys. Chem..‑ 1897.‑ 22.‑ P. 289; б) Лифшиц Е. М., Питаевский Л. П. Физическая кинетика. М.: Наука. Гл. ред. физ.-мат. лит., 1979.‑ 528 с.
- а) Лурье А. А. К теории колец Лизеганга / А. А. Лурьє // Коллоид, 1966.‑ 28(4.1).‑ C. 534-537; б) Полежаев А. А. Теория структур Лизеганга / А. А. Полежаев // «Математика. Компьютер. Образование». Сб. трудов X международной конференции. Под общей редакцией Г. Ю. Ризниченко Ижевск: Научно-издательский центр «Регулярная и хаотическая динамика», 2003.‑ Том 2.‑ С. 307-319.
- Равич-Щербо М. И. Новиков В. В. Физическая и коллоидная химия.‑М: Высшая школа, 1975.‑ 255 с.
- Bettelheim F. A., Landesberg J. M. / Laboratory Experiments for General, Organic, and Biochemistry, 6th ed.Thomson Brooks/Cole: Belmont, CA.‑ 2007.‑ P. 53−59.
- Vernon A. A. Treatment of Aluminum for Corrosion Prevention / A. A. Vernon // J. Chem. Educ.‑ 1949.‑ 26.‑ P. 147-148.
- Grotz L. C. Passivity of Aluminum / L. C. Grotz // J. Chem. Educ.‑ 1974.‑ 51.‑ P. 178.
- McCafferty E. Sequence of steps in the pitting of aluminum by chloride ions / E. McCafferty // Corrosion Science.‑ 2003.‑ 45 (7).‑ P. 1421-1438.
- Wei W. Y., Lee C., Chen H. J. Langmuir.- – 10.- P. 1980–1986.
Редакція може не поділяти думку авторів і не несе відповідальність за достовірність інформації. Будь-який передрук матеріалів з сайту може здійснюватись лише при наявності активного гіперпосилання на e-kolosok.org, а також на сам матеріал!




